HGDiscovery Analysis of HGD mutations in Alkaptonuria
Malancha Karmakar, Vittoria Cicaloni, Carlos H.M. Rodrigues, Ottavia Spiga, Annalisa Santucci & David B. Ascher
What is Alkaptonuria? Alkaptonuria (AKU) is a rare genetic metabolic disorder characterized by the accumulation of homogentisic acid (HGA) in the body. AKU patients may have dark urine or urine that turns black when exposed to air. Aside from dark urine that is present from infancy, affected alkaptonuric patients do not develop symptoms (asymptomatic) during infancy or childhood and often remain unaware of their condition until adulthood. They eventually develop ochronosis, which is the bluish-black discoloration of connective and other tissue within the body, and they may develop discoloration of the skin overlying cartilage within the body such as over part of the outer ear. In some cases, the whites of the eyes (sclera) may also become discolored. In adulthood, AKU patients also develop progressive arthritis of the spine and large joints.
What is HGD? Affected individuals lack enough functional levels of an enzyme required to breakdown HGA. Mutations in the HGD gene cause AKU and they are responsible for deficient levels of functional homogentisate 1,2-dioxygenase (HGD), which, in turn, leads to excess levels of HGA. Although HGA is rapidly cleared from the body by the kidneys, it also slowly accumulates in the various tissues of the body, especially connective tissue such as cartilage. Over time (rarely before adulthood), it eventually changes the color of affected tissue to a slate blue or black. Long-term, chronic accumulation of homogentisic acid eventually weakens and damages affected tissue and leads to many of the characteristic symptoms of AKU. Here we report a comprehensive mutation analysis of 111 HGD missense mutations.
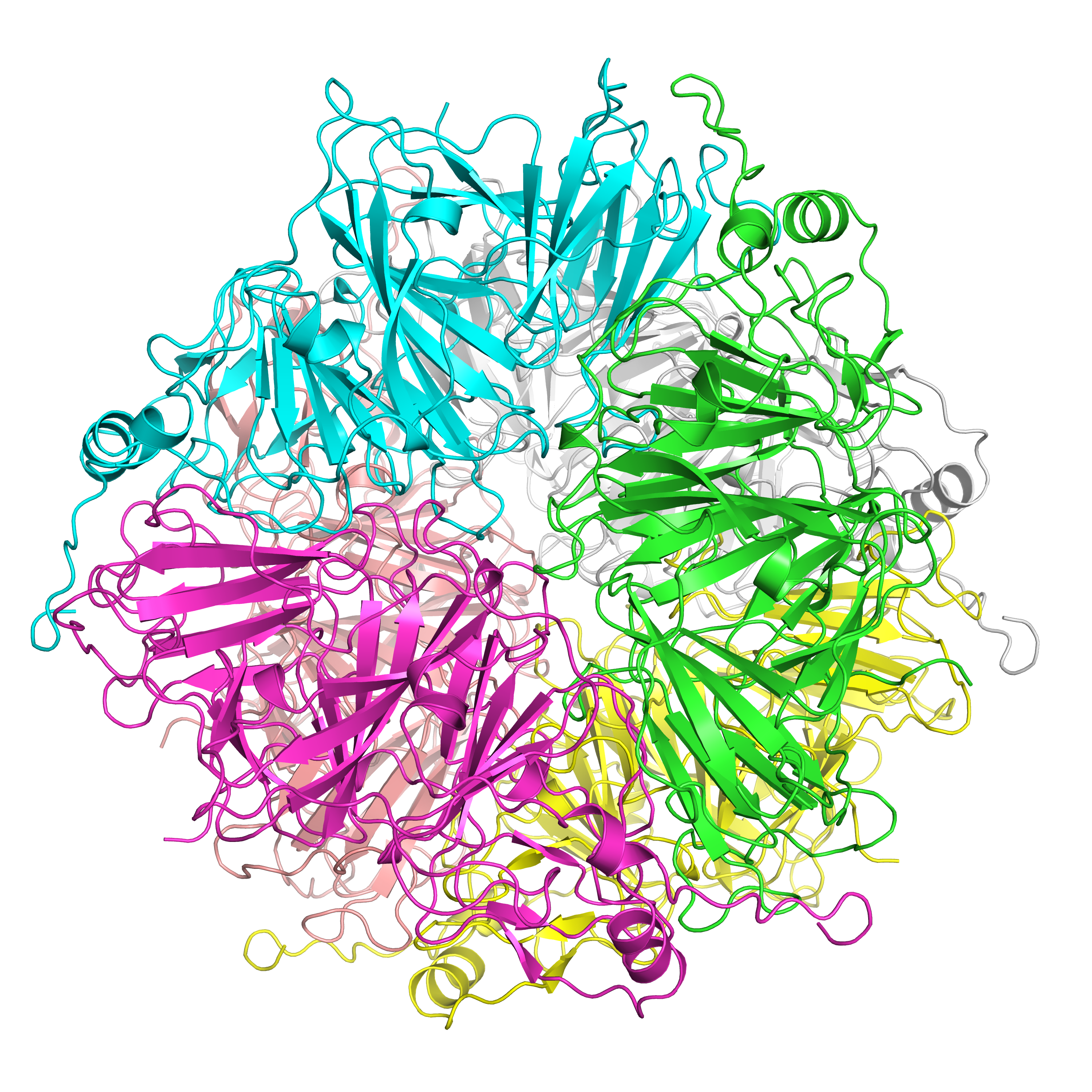
Karmakar, M., Cicaloni, V., Rodrigues, C.H.M., Spiga, O., Santucci, A., Ascher, D.B. (2022). HGDiscovery: an online tool providing functional and phenotypic information on novel variants of homogentisate 1,2- dioxigenase. Current Research in Structural Biology. Article in Revision.
Read manuscript description