DDMut provides a comprehensive suite for protein stability analysis.
Our deep learning method integrates our optimised graph-based signatures with other physicochemical properties to estimate the impact of point mutations on protein stability.
Running predictions (Single-point mutations)
Input
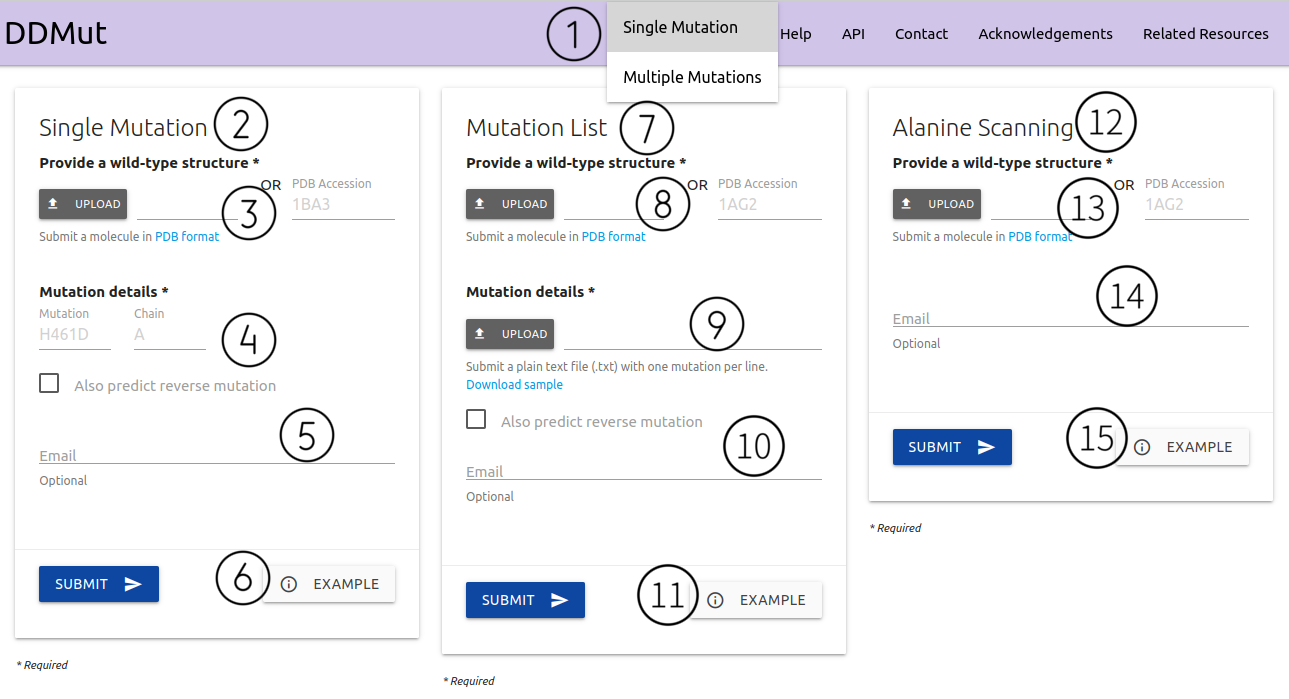
The input page for single-point mutations can be accessed from the home page or via the top menu Run > Single Mutation (1). Here, two options are available:
-
Single Mutation (2) - Submitting one single point mutation
- A structure must be provided as a file in PDB format or via PDB four letter accession code (3)
- Mutation details are required to be provided in the form of wild-type code + residue position + mutant code (using the one letter amino acid code)(4). Chain identifier is also required. There is a checkbox to also predict reverse mutations, but it will take a bit longer to run.
- If provided, an email will be sent to the user after the submission is processed (5)
- Examples of inputs format and results are also available (6)
-
Mutation List (7) - Submitting a list of single point mutations to be analysed and processed in batch
- Like the Single Mutation option, here users are also required to input a structure must be provided as a file in PDB format or via PDB four letter accession code (8)
- Mutation details are required to be provided as a plain text file (preferable format is .txt). Each mutation must be defined as chain identifier + blank space + wild-type code + residue position + mutant code (using the one letter amino acid code) (9). A sample file is available for download. There is a checkbox to also predict reverse mutations, but it will take a bit longer to run.
- If provided, an email will be sent to the user after the submission is processed (10)
- Examples of inputs format and results are also available (11)
-
Alanine Scanning (12) - Submitting a structure and mutating every single residue to Alanine
- A structure must be provided as a file in PDB format or via PDB four letter accession code (13)
- If provided, an email will be sent to the user after the submission is processed (14)
- Examples of inputs format and results are also available (15)
Results
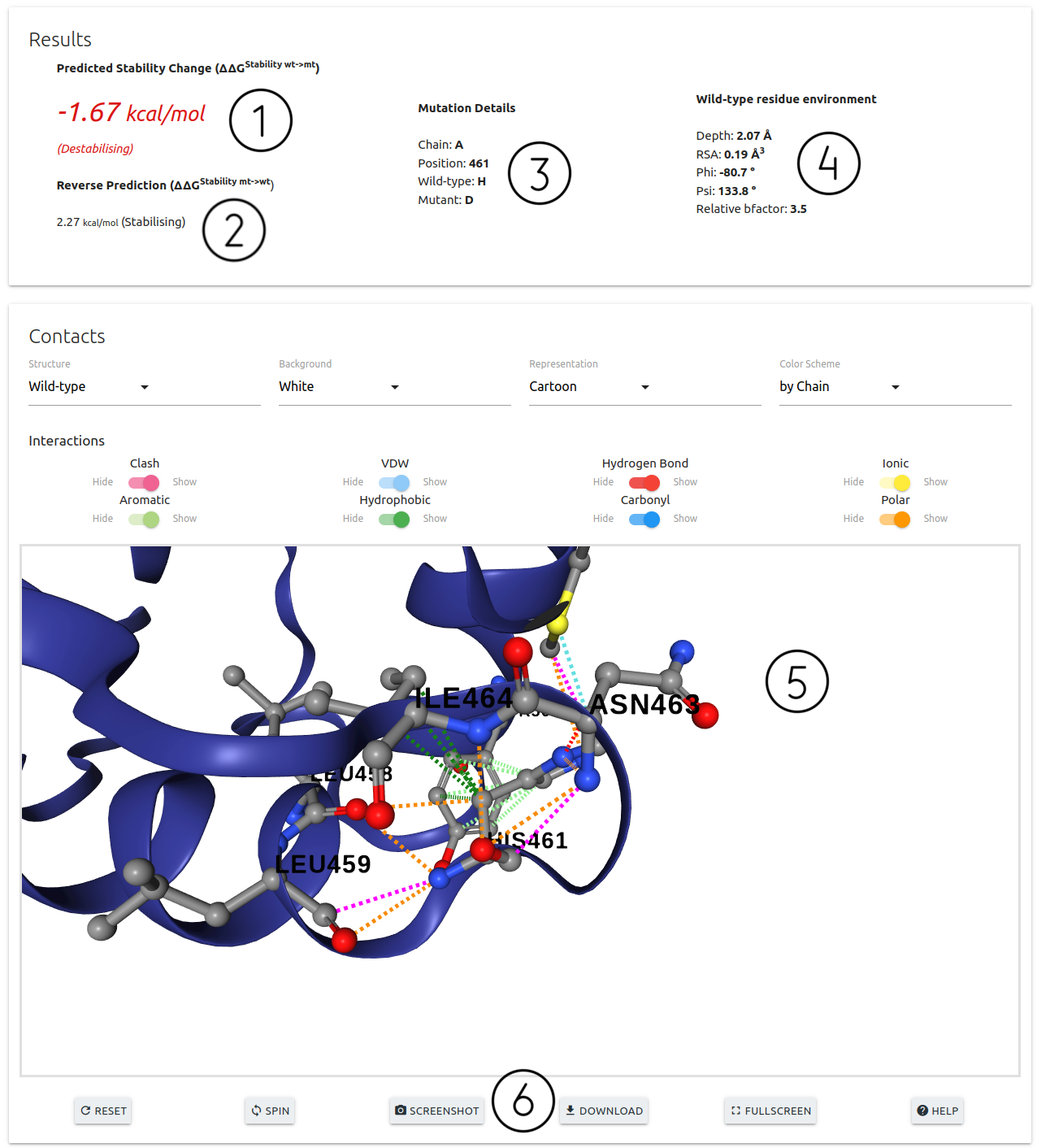
On the results page for Single Mutation option:
- DDMut predictions for the forward (1) and reverse (2) mutations are shown on the top section of this page alongside with details on the input mutation (3) and wild-type residue environment (4)
- An interactive 3D viewer is available (5) allowing for the analysis of interatomic interactions for the wild-type residue. A set of controls are also available for customising the viewer
- Action buttons at the bottom of the viewer (6)
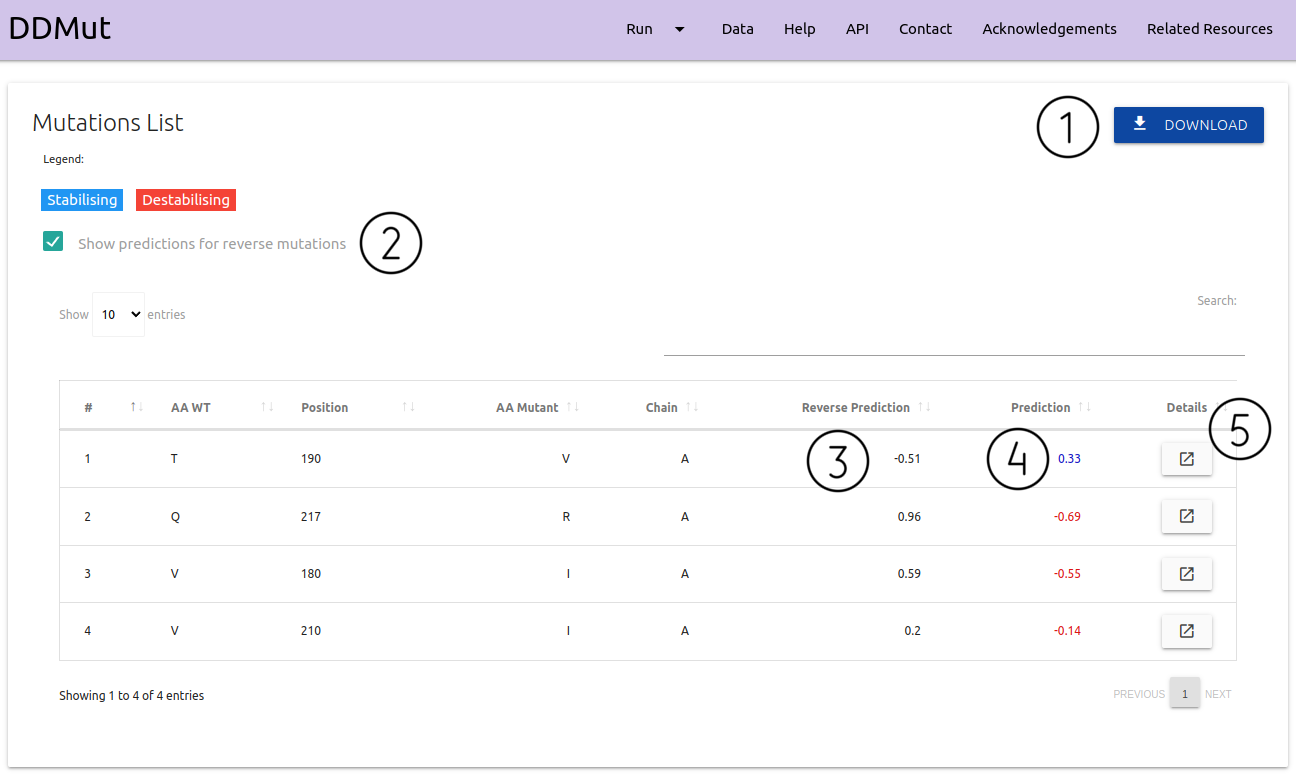
For Mutation List option:
- Results are summarised as a downloadable table (1)
- Details on the input mutations and predictions (4) for each entry are shown
- There is a checkbox (2) to show and hide the column for reverse predictions (3) if the user chose to also predict these
- Each entry can be visualised individually via the Details (5) button
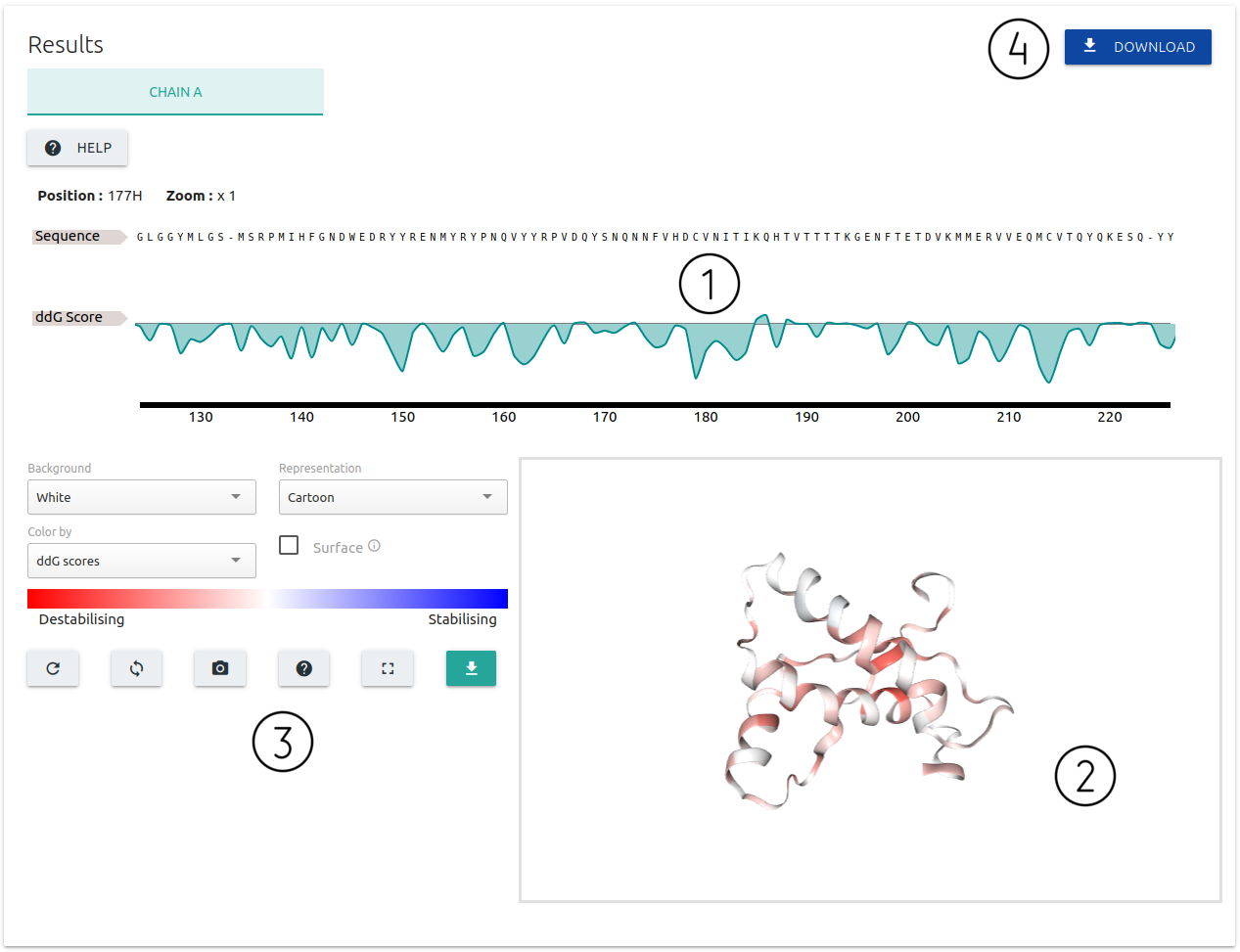
For Alanine Scanning option:
- Results are summarised in relation to the sequence of amino acids present in each chain of the submitted protein structure (1), and also as an interactive 3D viewer (2) where scores for each residue are mapped onto the input structure
- A set of controllers and action buttons are available for customising the 3D viewer (3)
- There is a button to download the results as a table (4)
Running predictions (Multiple mutations)
Input
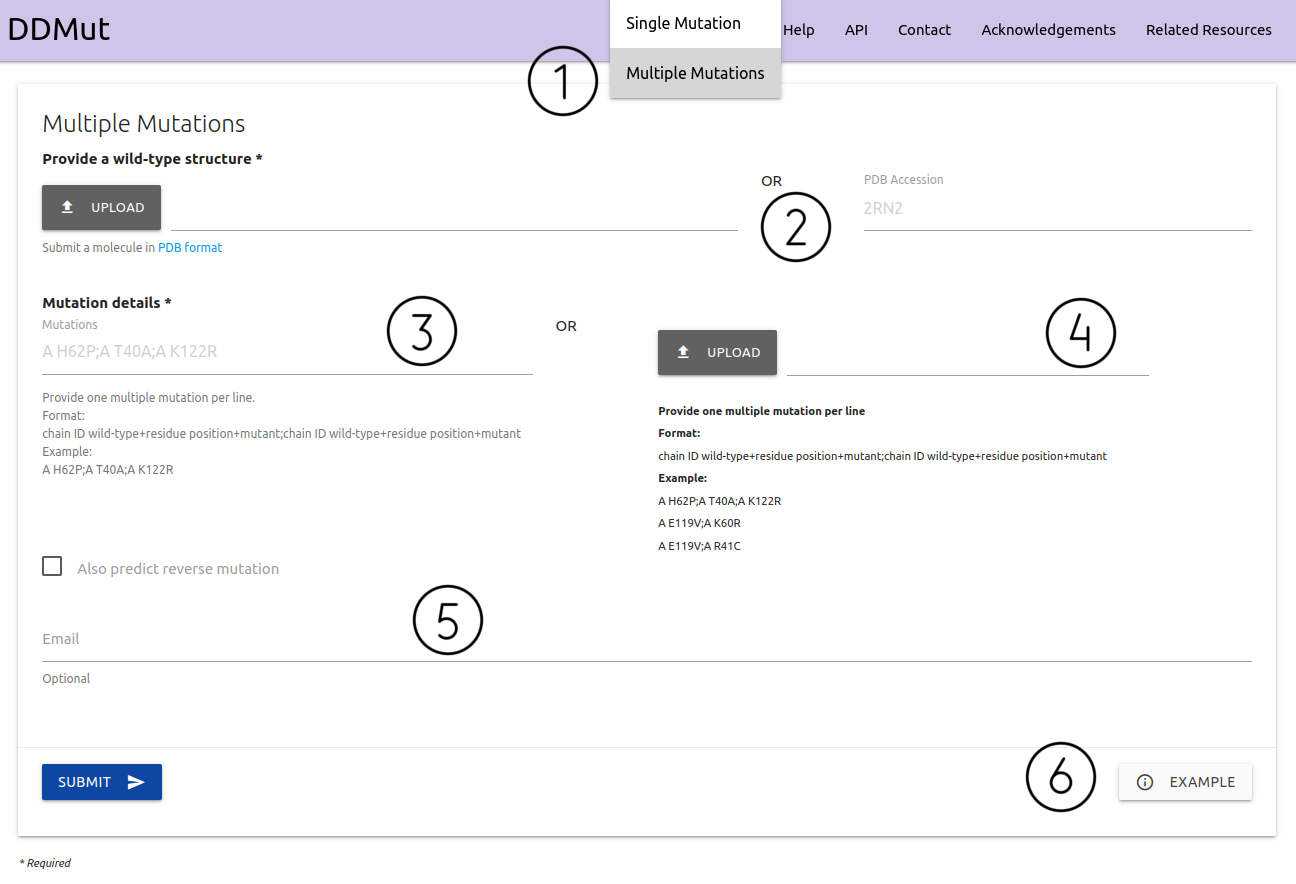
The input page for running predictions for multiple mutations (double and triple mutants) is available from the home page or via the top menu Run > Multiple Mutations (1)
- A structure must be provided as a file in PDB format or via PDB four letter accession code (2)
- Users can input a single entry (3) or a list of multiple mutations to be processed in batch (4). Entries are required to be provided in the form of Chain identifier + blank space + wild-type code + residue position + mutant code (using the one letter amino acid code) separated by a semi-colon (;). Example inputs are available. There is a checkbox to also predict reverse mutations
- If provided, an email will be sent to the user after the submission is processed (5)
- Examples of inputs format and results are also available (6)
Results
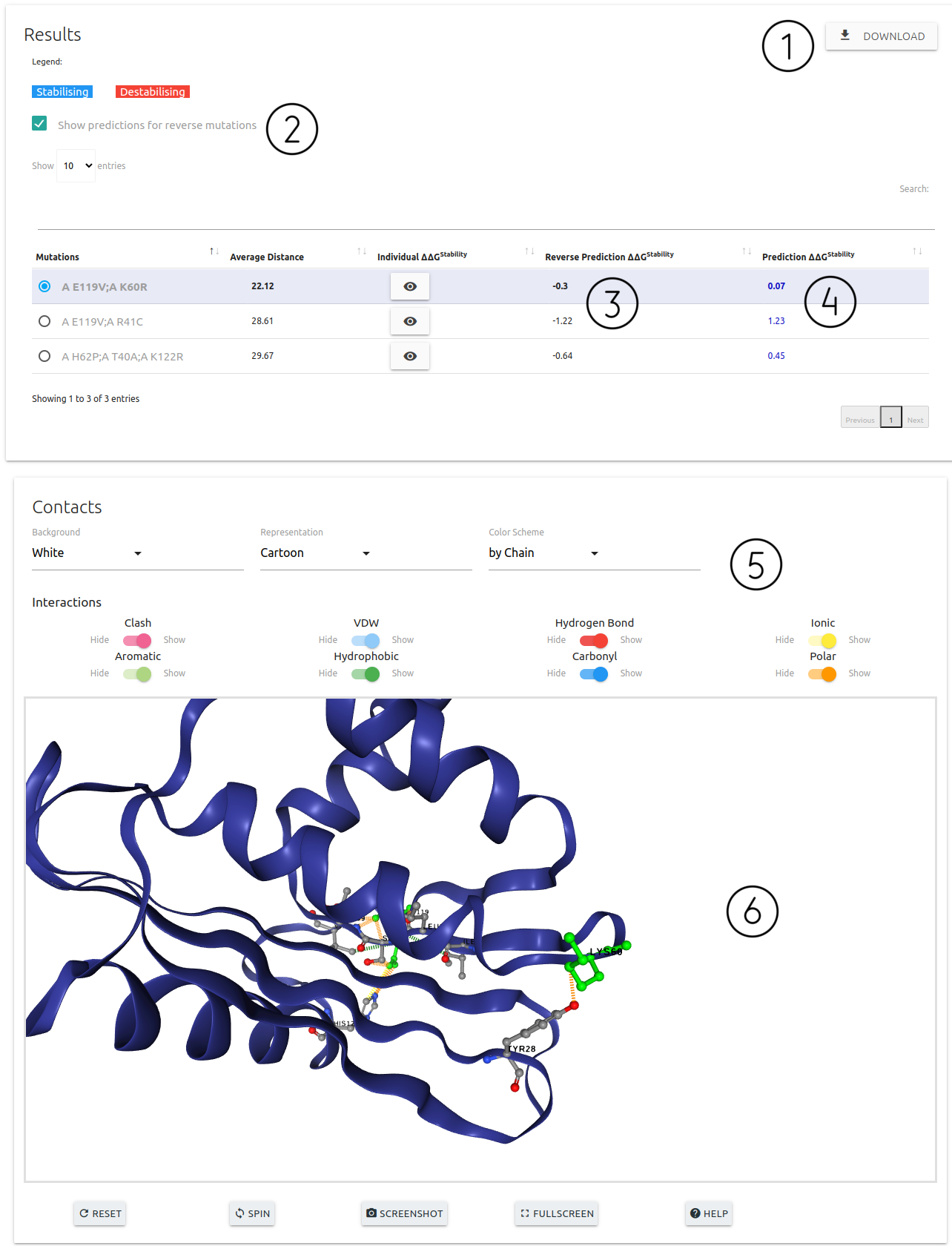
- Results are summarised as a downloadable table (1). Each entry on the table can be selected to be displayed on the 3D viewer (6)
- Details on the input mutations and predictions (4) for each entry are shown. Entries predicted as stabilising are shown in blue and destabilising in red. Individual ΔΔGStability for each point mutation can also be viewed by clicking the button visibility. These are calculated using DDMut Single Mutation model.
- There is a checkbox (2) to show and hide the column for reverse predictions (3) if the user chose to also predict these
- A set of controls are available for customising the viewer (5)
Running Normal Mode Analysis
Input
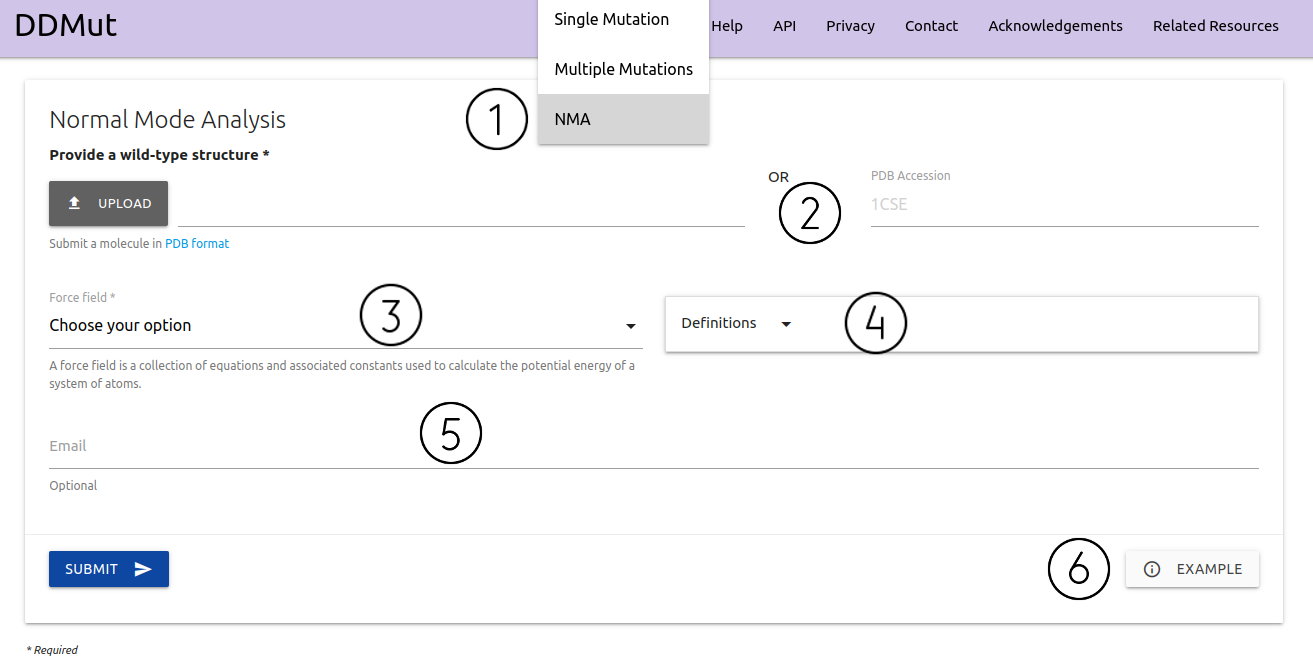
Normal Mode Analysis is available from the home page or via the top menu Run > NMA (1)
- Similarly to running predictions, here users are also required to provided a file in PDB format or a PDB four letter accession code (2)
- A force field must also be provided (3). The Normal Mode Analysis available here are based on the bio3D package that uses the C-alpha force field by default. However, no specific force field is strictly recommend. A summary of options available with a brief description and references is shown in (4).
- If provided, an email will be sent to the user after the submission is processed (5)
- Examples of inputs format and results are also available (6)
Results
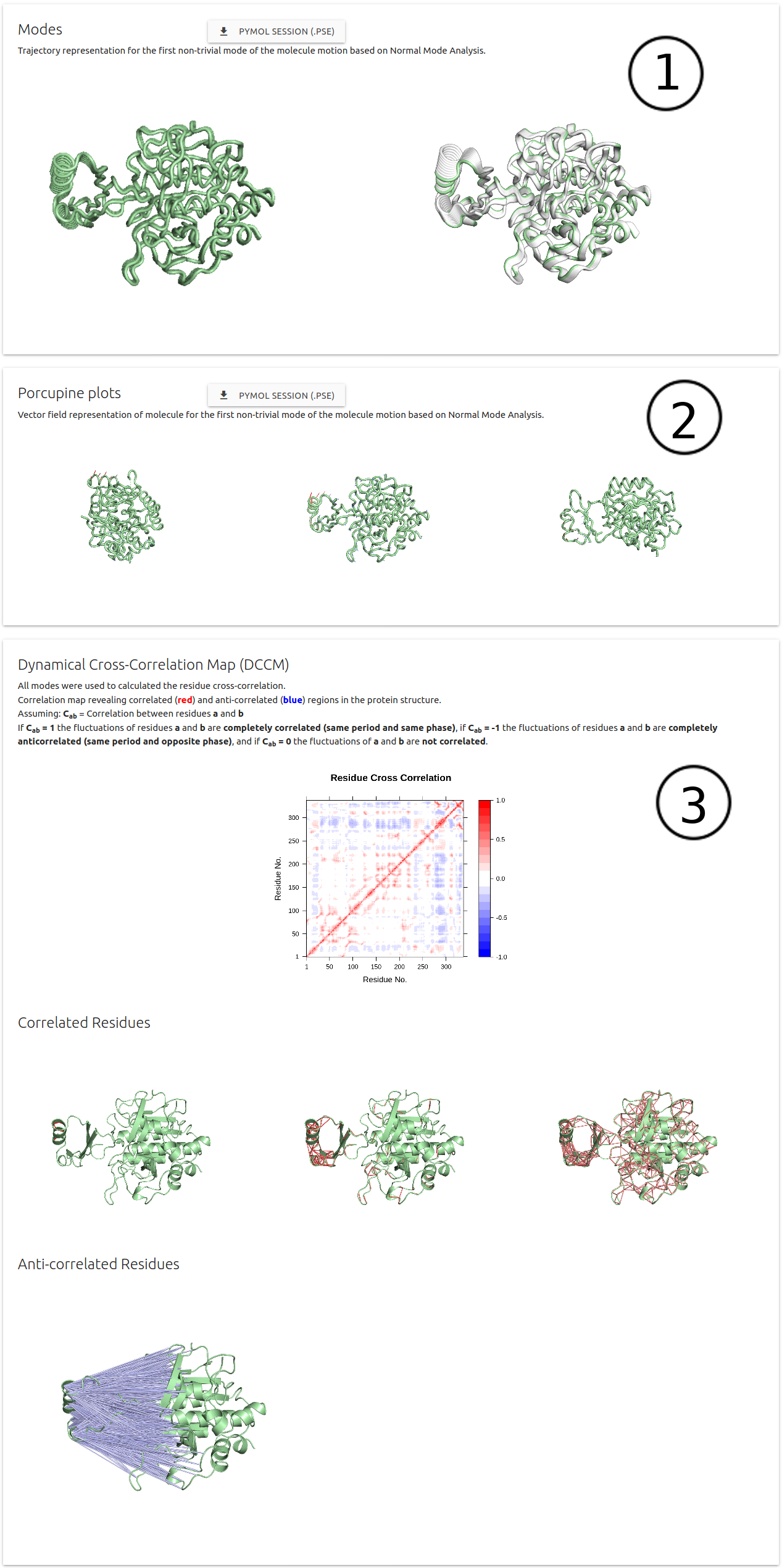
NMA results are divided in 3 sections:
- (1) Modes: Trajectory representation for the firt non-trivial mode of the molecule motion.
- (2) Porcupine plots: Vector field representation of molecule for the first non-trivial mode of the molecule
- (3) Dynamical Cross-Correlation Map (DCCM): Residue cross-correlation using all modes
- Pymol session files are available for download
Contact us
In case you experience any troube using DDMut or if you have any suggestions or comments, please do not hesitate in cantacting us either via email or via our Group website.
If your are contacting regarding a job submission, please include details such as input information and the job identifier