DDMut Predicting mutation effects on protein stability using deep learning
Yunzhuo Zhou, Qisheng Pan, Douglas E.V. Pires, Carlos H.M. Rodrigues & David B. Ascher
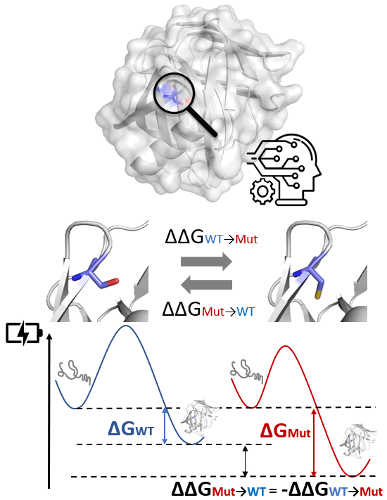
Abstract: Understanding the effects of mutations on protein stability is crucial for variant interpretation and prioritisation, protein and cell design, engineering, and biotechnology. While experimental measurements are the gold-standard, they can be time consuming, expensive and technically challenging. The data generated from these approaches, however, has driven an explosion in computational approaches to predict the effects of mutations on protein stability. Despite these efforts, community assessments of these tools have highlighted a range of limitations, including availability, high time costs, low predictive power, and biassed predictions towards destabilising mutations. To fill this gap, we developed DDMut, a fast and accurate siamese network to predict changes in Gibbs Free Energy (ΔΔG) upon single and multiple point mutations, leveraging both forward and hypothetical reverse mutations to account for model anti-symmetry. Deep learning models were built by integrating graph-based representations of the localised 3D environment, with convolutional layers and transformer encoders. This combination could better capture the distance patterns between atoms by extracting both short-range and long-range interactions. DDMut achieved pearson's correlation of up to 0.70 (RMSE: 1.37 kcal/mol) on predicting single point mutation on cross-validation, and 0.74 (RMSE: 1.67 kcal/mol) on multiple mutations, and outcompeted most available methods on non-redundant blind test sets. Importantly, DDMut was highly scalable and demonstrated anti-symmetric performance on both destabilising and stabilising mutations. We believe DDMut will be a useful platform to better understand the functional consequences of mutations, and guide rational protein engineering.
This website is free for all users. For  policy please click here.
policy please click here.