MTR-Viewer
Missense Tolerance Ratio: Identifying regions within genes under purifying selection.
Michael Silk, Slavé Petrovski, David B. Ascher.
Abstract
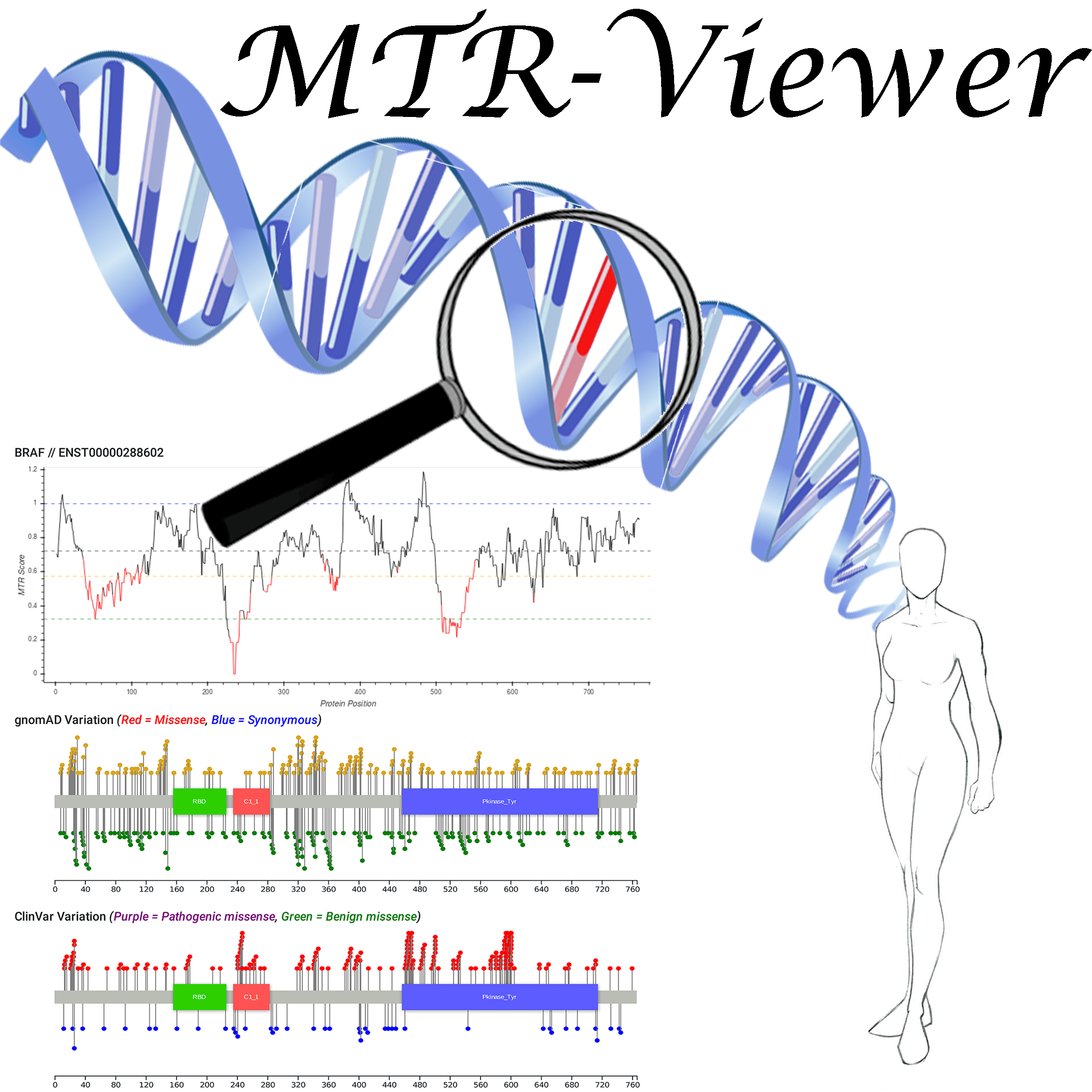
Motivation: Advances in genomic sequencing have enormous potential to revolutionise personalised medicine, however distinguishing disease-causing from benign variants remains a challenge. The increasing number of human genome and exome sequences available has revealed areas where unfavourable variation is removed through purifying selection. Here we present the MTR-Viewer, a web-server enabling easy visualisation at the gene or variant level of the Missense Tolerance Ratio (MTR), a measure of regional intolerance to missense variation calculated using variation from 220,000 exome and genome sequences. The MTR-Viewer enables exploration of MTR calculations, using different sliding windows, for over 18,000 human protein-coding genes and 85,000 alternative transcripts. Users can also view MTR scores calculated for specific ethnicities, to enable easy exploration of regions that may be under different selective pressure. The spatial distribution of population and known disease variants is also displayed on the protein’s domain structure.
Results: Intolerant regions were found to be highly enriched for ClinVar pathogenic and COSMIC somatic missense variants (Mann-Whitney U test p < 2.2x10-16). As the MTR is not biased by known domains and protein features, it can highlight functionally important regions within genes overlooked or inaccessible by traditional methods. MTR-Viewer is freely available via a user friendly web-server at /mtr-viewer/
| View the MTR scores for your gene or transcript | |
| Look up the MTR scores for your set of variants |
Traynelis J.,* Silk M.,* Wang Q., Berkovic S.F., Liu L., Ascher D.B., Balding D.J., Petrovski S. (2017). Optimizing genomic medicine in epilepsy through a gene-customized approach to missense variant interpretation. Genome Research