About
What is the Missense Tolerance Ratio?
The MTR aims to quantify the amount of purifying selection acting specifically on missense variants in a given window of protein-coding sequence. It is estimated across sliding windows of 31 codons (default) and uses observed standing variation data from the WES component of gnomAD / the Exome Aggregation Consortium Database (ExAC), version 2.0 (http://gnomad.broadinstitute.org).
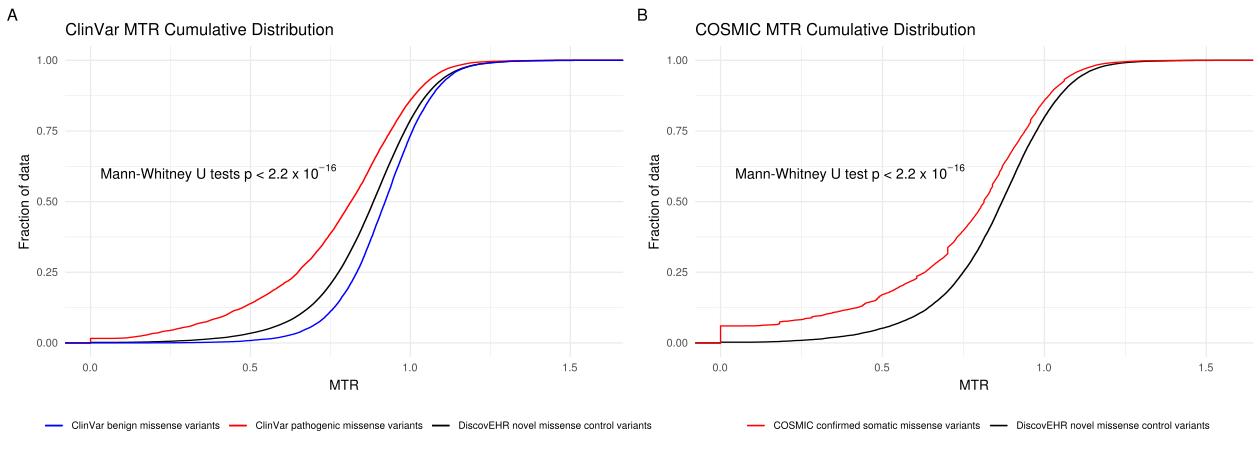
Distribution of MTR scores for known disease variants compared to background. (A) Cumulative distribution of MTR scores for ClinVar pathogenic variants (red), ClinVar benign variants (blue) and DiscovEHR novel missense control variants (black). (B) Cumulative distribution of MTR scores for COSMIC somatic missense variants (red) compared with DiscovEHR novel missense control variants (black).
How are the MTR scores calculated?
Please refer to the associated publication, Traynelis J.,* Silk M.,* Wang Q., Berkovic S.F., Liu L., Ascher D.B., Balding D.J., Petrovski S. (2017). Optimizing genomic medicine in epilepsy through a gene-customized approach to missense variant interpretation. Genome Research (2017).
Lollipop Plots of Variation
Lollipop plots are given for the gnomAD variation for a given gene, where there i sa valid HGNC symbol and Pfam domain representation for that symbol. If the HGNC symbol matches to a gene in ClinVar's set of disease-associated genes, a lollipop plot is given for the non-ambiguous benign and missense variants found.
These are adapted from the GitHub project here: https://github.com/pbnjay/lollipops
Can I access the MTR scores via any batch processes?
The MTR scores are available via the Variant Effect Predictor (VEP) using a plugin at https://www.ensembl.org/info/docs/tools/vep/script/vep_plugins.html. This can be used to annotate variants with the MTR scores.
They can also be accessed using an API to query variants using a web-browser http://biosig.unimelb.edu.au/mtr-viewer/api/search or via command line using curl -i http://biosig.unimelb.edu.au/mtr-viewer/api/search, where "search" allows for identical query formatting as in the queries page.
Querying via the API returns a JSON object with the same columns each time, and can be used for easy pipeline integration.
How do I use the MTR-Viewer?
Enter a gene symbol (HGNC), Ensembl transcript ID (v75) or corresponding Refseq transcript ID into the search bar. The viewer will display the gene name and Ensembl Transcript ID. If a gene symbol (HGNC) is supplied, the viewer will default to a canonical transcript of our choosing. Alternate transcripts are listed below the viewer.
1: Quickly query a gene or variant with the provided search box from any page
2: Additional search box on homepage. Alternatively, you can select to run an example.
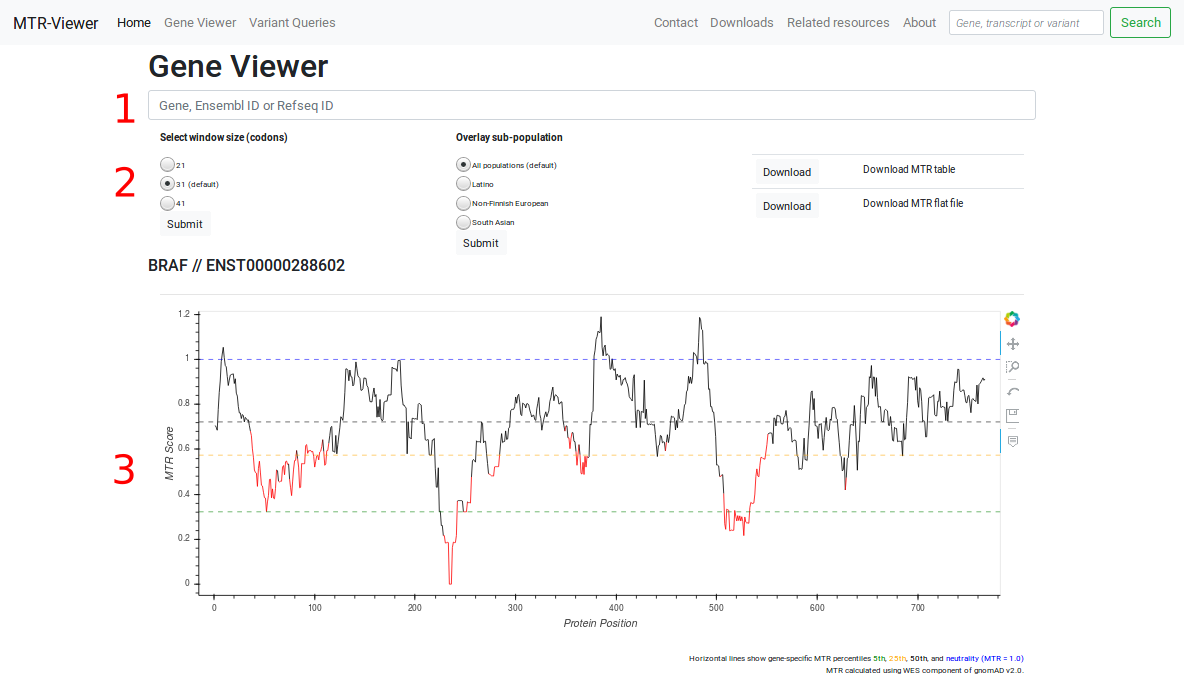
1: Query a gene, Ensembl transcript (v75) or Refseq transcript here. If querying an HGNC symbol, defaults to either the canonical-defined Ensembl transcript or the longest CCDS available.
2: Select window size between 21 31, and 41 codon MTR calculations. A within-population MTR calculation can be overlaid also for populations with at least 15,000 exomes in the input dataset. Population overlays are not available for 21-codon windows due to insufficient sample sizes to examine the MTR within a population for regions this small. Download links allow for downloading transcripts in a table format (one MTR score per protein position) or flat file for default 31 codon windows (MTR scores given to each genomic variant within a transcript).
3: The MTR distribution for a gene is plotted across the protein-coding sequence (x-axis showing protein / codon position). Low-MTR scores indicate stronger purifying selection of missense variants. Line sections are coloured red where the FDR-adjusted binomial exact test < 0.1, quantifying MTR deviation from neutrality. (MTR = 1). Horizontal lines show summary statistics for the displayed gene's MTR scores (green: 5th %ile, orange: 25th %ile, black: median, blue: MTR = 1). MTR is not calculated where a region has fewer than 3 variants, shown as gaps in the line plot.
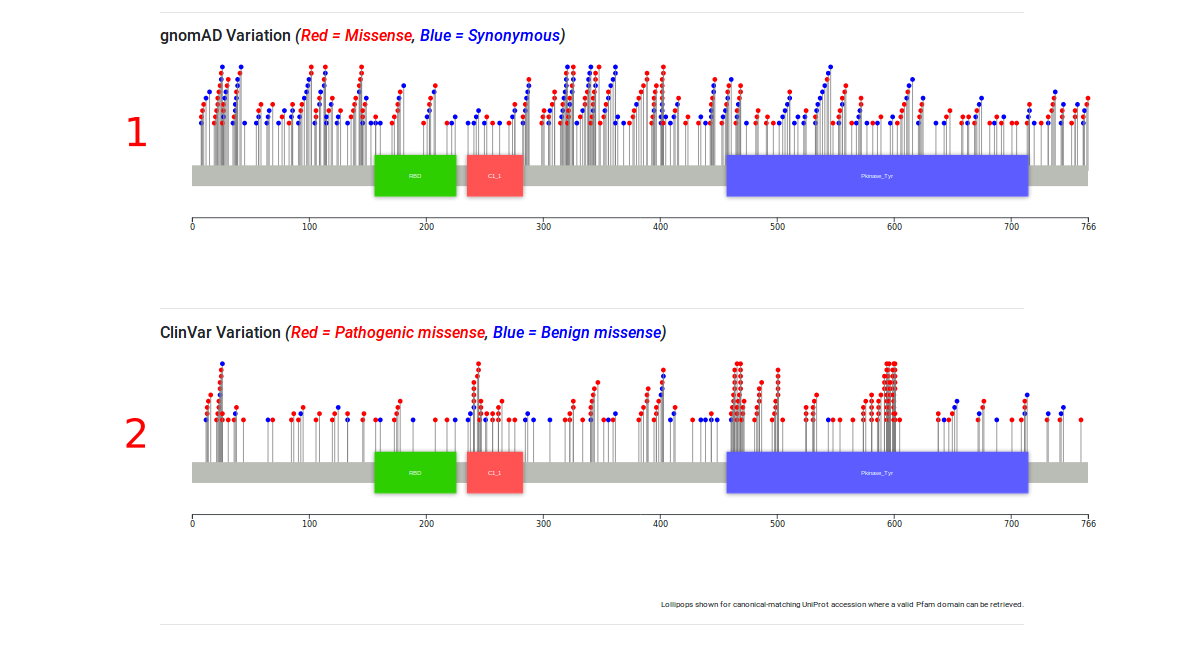
1: Lollipop plot showing the observed gnomAD synonymous (blue) and missense (red) variation. Protein domains are sourced for Pfam and as such, lollipops are only displayed for the canonical transcript with an available UniProt accession.
2: Lollipop plot showing ClinVar pathogenic missense (red) and benign missense (blue) variants. Variants are filtered to only those with no conflicting evidence in ClinVar.
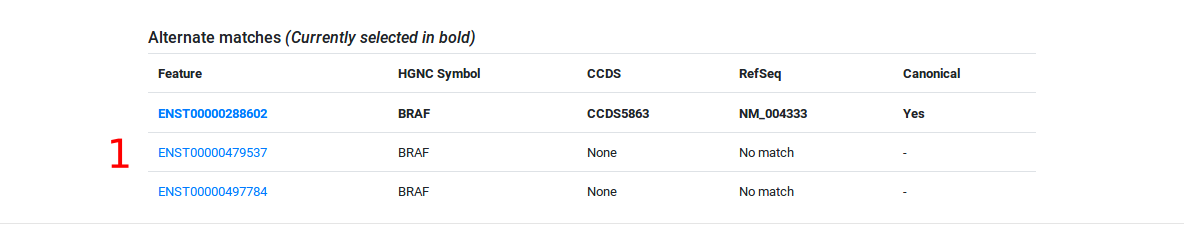
1:Available transcripts related to your current search are shown here. CCDS and RefSeq names are shown here if listed in Ensembl v75. Current search is highlighted in bold.
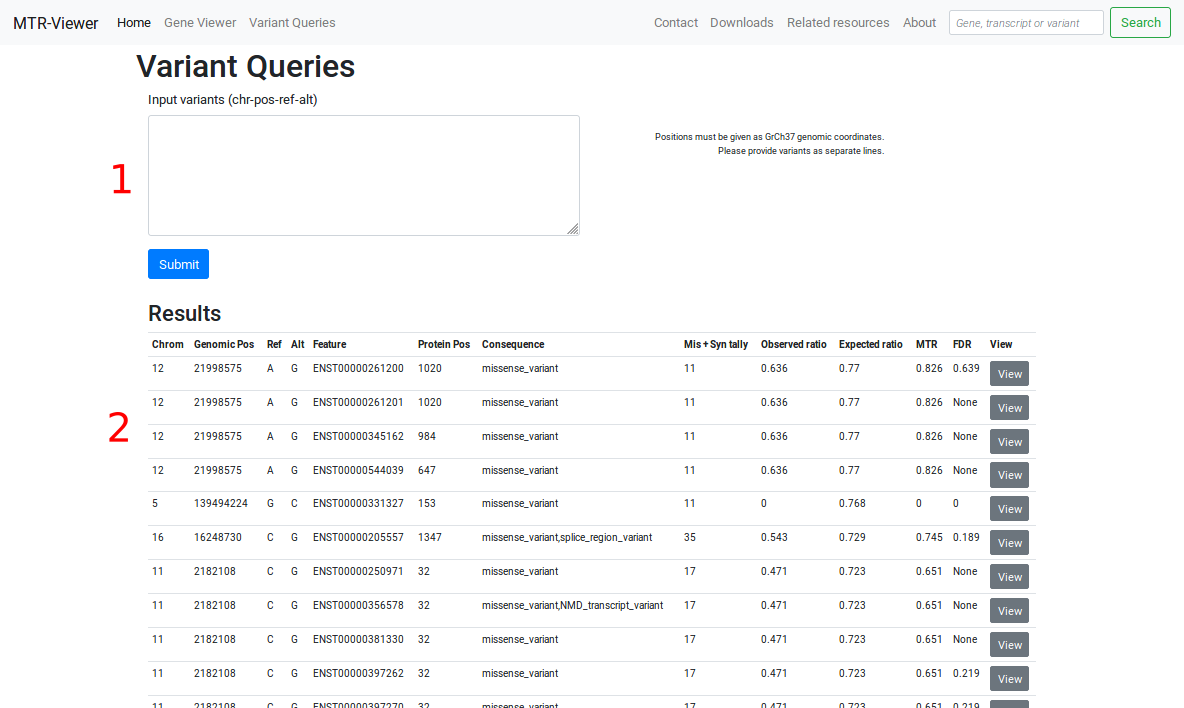
1: Input a list of variants in the form Chrom-Pos-Ref-Alt, each on a separate line. GrCh37 genomic coordinates are currently required.
2: Query results are shown for all matching transcripts for each variant. A link is provided to view the gene of a given match.