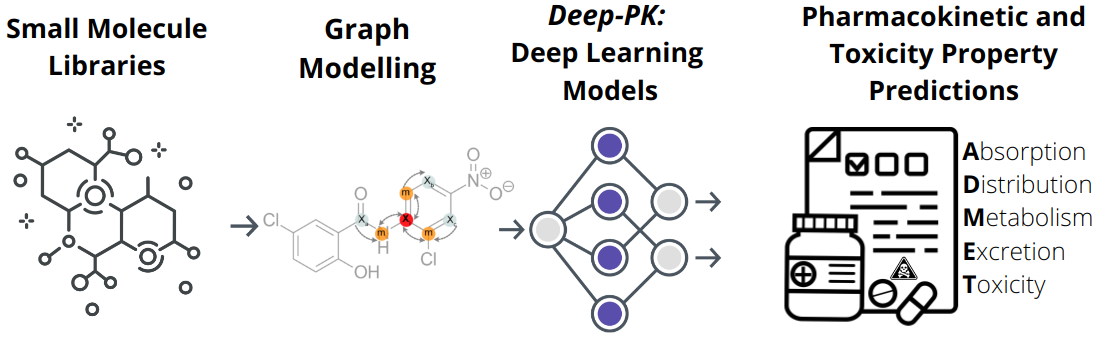
Deep-PK deep learning for small molecule pharmacokinetic and toxicity prediction
Abstract:
Evaluating the pharmacokinetic (PK) properties of small molecules is considered a key feature in most drug development and high-throughput screening processes. Generally, PKs, which represent the fate of drugs in the human body, are described from four perspectives: Absorption, Distribution, Metabolism, and Excretion – all of which are closely related to a fifth perspective, Toxicity (ADMET). Since obtaining PK and toxicity data from in vivo or pre-clinical stages is time-consuming and expensive, many efforts have been made to predict ADMET properties via computational approaches. However, the majority of available methods are limited in their ability to provide PKs and toxicity for diverse targets, ensure good overall accuracy, and offer ease-of-use, interpretability, and extensibility for further optimisations. Here, we introduce Deep-PK, a deep learning-based PK and toxicity prediction, analysis and optimisation platform. We applied Graph Neural Networks and graph-based signatures as a graph-level feature to yield the best predictive performance across 73 endpoints including 64 ADMET and 9 general properties. With these powerful models, Deep-PK supports molecular optimisation and interpretation, aiding users in optimising and understanding PKs and toxicity for given input molecules.
Important Information:
- This website is free for all users.
- For cookie policy please click here.