Mutation Viewer
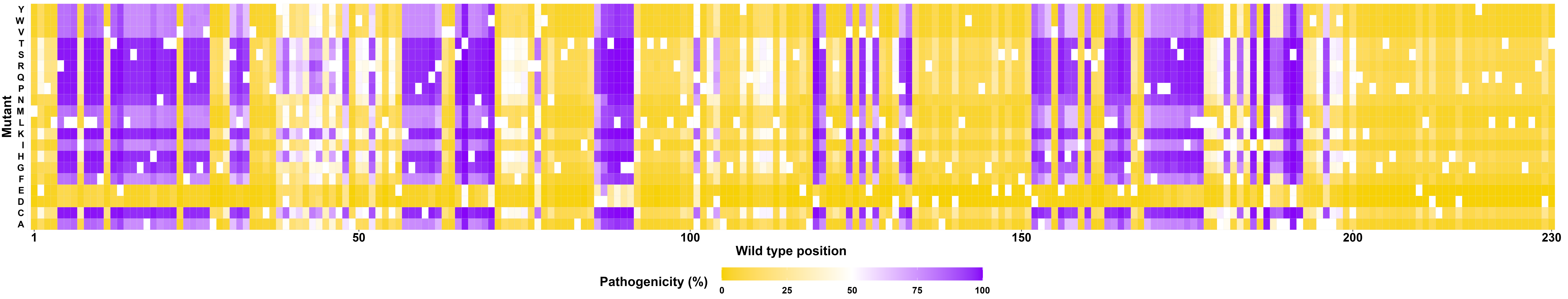
Please choose a missense mutation of interest from the heatmap, and click to get protein structural information.
Missense Mutation Selection
| Result |
Note |
- Wild-type
- Position
- Mutant
|
- Missense mutations are coordinated by the wild-type amino acid at the bottom (x-axis) and the mutant amino acid on the left (y-axis).
- Wild-type positions are based on canonical sequence.
|
| Prediction |
Predicted phenotypes |
| Confidence |
- Predicted probability of the phenotypes, showing how confidence the prediction is.
|
Residue Structural Environment
| Feature |
Explanation |
| Secondary Structure |
Secondary structure at the mutation site: Helix, Strand, or Loop. |
| Relative solvent accessibility (RSA) |
- This feature (range: 0-1) describes how the residue is exposed to solvent.
- A residue with a low RSA (e.g. < 20%) is usually located in the buried reigon, while a resiude with a high RSA (e.g. > 20%) is usually more exposed to solvent / closer to the surface.
|
| Phi & Psi |
Peptide torsion angles of the wild-type residue. |
| Predicted Local Distance Difference Test (pLDDT) |
- This feature (range: 0-100) describes how AlphaFold2 is confident with the predicted residue conformation.
- Residue with higher pLDDT score is considered as more accurately predicted, while the region with a low pLDDT score (low confidence) is often associated with disorderness.
- pLDDT > 90: Highly confident
- 90 > pLDDT > 70: Confident
- 70 > pLDDT > 50: Low confident
- 50 > pLDDT: Very low confident
- More information: click here
|
Mutation Effect
| Feature |
Explanation |
| Protein stability |
-
In-silico biophysical measurement of the change of protein stability (ΔΔG), computed by DDMut open_in_new.
-
We used 0 as a cutoff:
-
ΔΔG < 0 Kcal/mol means this mutation is destabilising the structure;
-
ΔΔG > 0 Kcal/mol means this mutation is stabilising the structure;
-
ΔΔG = 0 Kcal/mol refers to netural effect.
|
Position-specific substitution
matrix (PSSM) |
- Position-specific conservation, computed by PSI-BLAST.
- Higher score indicates that this amino acid is more conserved.
|
| AlphaMissense prediction |
- Variant effect predictor built on AlphaFold2 open_in_new.
- According to AlphaMissense, the cutoffs to classify variants are 0.34 and 0.564.
- AlphaMissense_score < 0.34 means this mutation is "likely_benign";
- AlphaMissense_score > 0.564 means this mutation is "likely_pathogenic";
- AlphaMissense_score in (0.34, 0.564) means this mutation is "ambigious" in phenotypes.
|
| ESM1b prediction |
- Prediction of disease variant effects using a deep protein language model open_in_new.
- There is no specific cutoff to define pathogenic variants.
- But, the smaller the ESM1b_score is, the more likely this variant can be disease-causing.
|
| CPT-1 prediction |
- A cross-protein transfer model to annotate disease variants open_in_new.
- There is no specific cutoff to define pathogenic variants.
- But, the larger the CPT1_score is, the more likely this variant can be disease-causing.
|
{kind=link}