SUSPECT-BDQ StrUctural Susceptibility PrEdiCTion on BDQ
Malancha Karmakar, Carlos H.M. Rodrigues, David B. Ascher & Justin Denholm
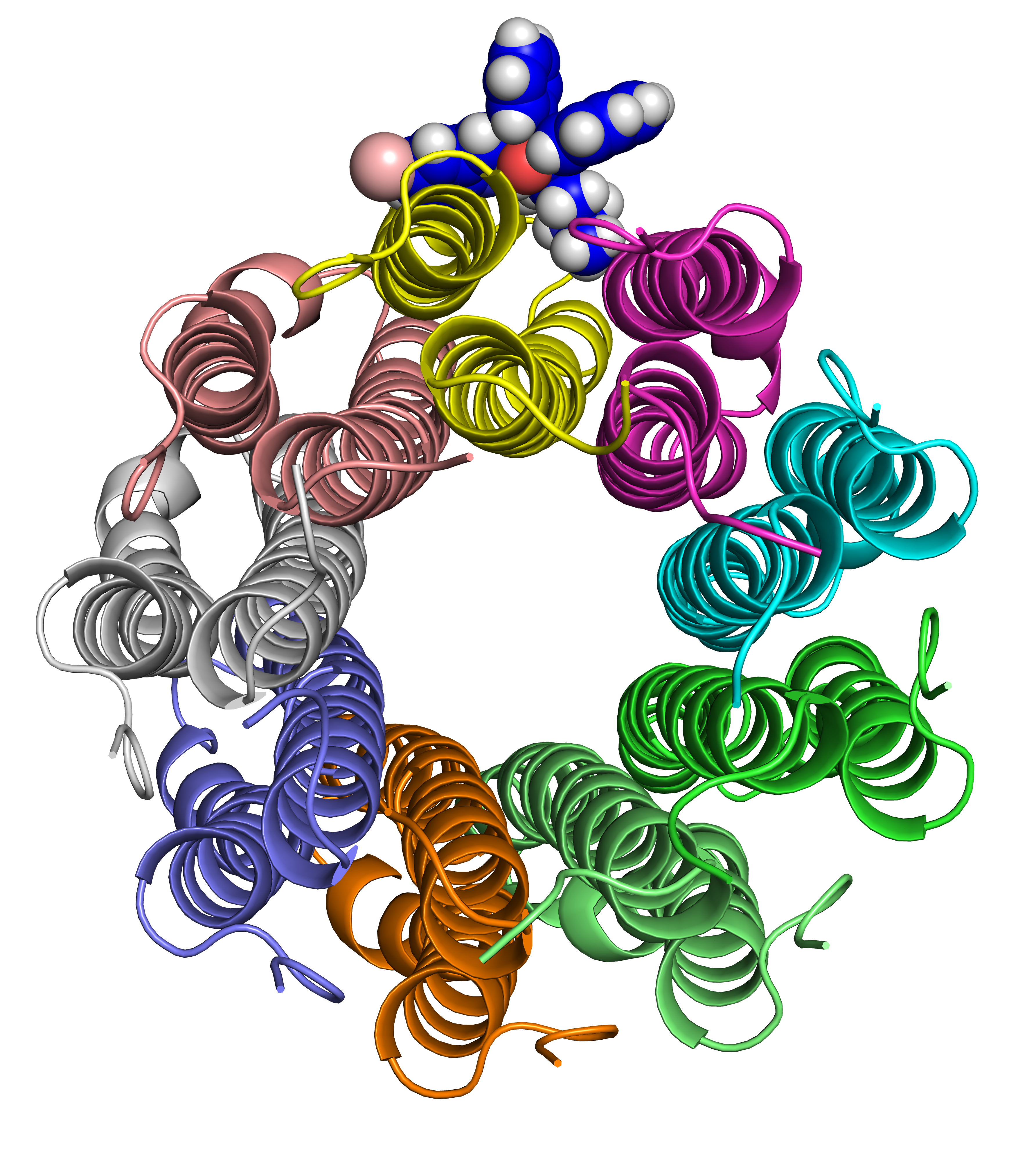
Abstract: Clinical resistance against Bedaquiline, the first new anti-tuberculosis compound with a novel mechanism of action in over 40 years, has already been detected in Mycobacterium tuberculosis. As a new drug, however, there is currently insufficient clinical data to facilitate reliable and timely identification of genomic determinants of resistance. Here we investigate the structural basis for M. tuberculosis associated bedaquiline resistance in the drug target, AtpE. Together with the 9 previously identified resistance-associated variants in AtpE, 54 non-resistance-associated mutations were identified through comparisons of bedaquiline susceptibility across 23 different mycobacterial species. Computational analysis of the structural and functional consequences of these variants revealed that resistance associated variants were mainly localized at the drug binding site, disrupting key interactions with bedaquiline leading to reduced binding affinity. This was used to train a supervised predictive algorithm, which accurately identified likely resistance mutations (93.3% accuracy). Application of this model to circulating variants present in the Asia-Pacific region suggests that current circulating variants are likely to be susceptible to bedaquiline. This tool could be useful for the rapid characterization of novel clinical variants, to help guide the effective use of bedaquiline, and to minimize the spread of clinical resistance. We have made this model freely available through a user friendly web interface called SUSPECT-BDQ, StrUctural Susceptibility PrEdiCTion for bedaquiline. This will be a valuable resource to analyse any atpE missense mutation, providing structural insight to help guide patient treatment decisions and screening programs.